
Превращение фенольных соединений
Превращение фенольных соединений
Характеристика фенольных соединений
Фенольные соединение – вещества ароматической природы, которые содержат одну или несколько гидроксильных групп, связанных с атомами углерода ароматического ядра.
Фенольные соединения представляют собой один из наиболее распространенных и многочисленных классов природных соединений, обладающих биологической активностью. Фенольные соединения наиболее распространены и свойственны практически каждому растению и даже каждой растительной клетке.
Отличной чертой фенольных соединений является формирование огромного числа соединений за счет модификаций молекулы и образования конъюгатов с разнообразными структурами. Из модификаций для фенольных соединений характерны образование глюкозидов, метилирование и метоксилирование. За счет гидроксильных и карбоксильных групп фенольные соединения могут связываться с сахарами, органическими кислотами, растительными аминами, алкалоидами. Помимо этого растительные фенолы могут соединяться с изопреноидами, образуя большую группу пренилированных фенолов. Такие свойства фенольных соединений обеспечивают огромное разнообразие структур, характерное для растительных фенолов.
По числу ОН-групп различают одноатомные (например, сам фенол), двухатомные (пирокатехин, резорцин, гидрохинон) и многоатомные (пирогаллол, флороглюцин и др.) фенольные соединения.
Фенольные соединения существуют в свободном виде и виде мономеров, димеров, олигомеров и полимеров, в основу классификации природных фенолов положен биогенетический принцип. В соответствии с современными представлениями о биосинтезе их можно разбить на несколько основных групп:
соединения С6-ряда – простые фенолы, бензохиноны;
соединения С6 – C1-ряда – производные бензойной кислоты (фенольные кислоты);
соединения С6 – C2-ряда – фенолспирты и фенолуксусные кислоты, ацетофеноны;
соединения С6 – C3-ряда – производные фенилпропана (оксокоричные кислоты и спирты, фенилпропены, кумарины, изокумарины и хромоны);
соединения С6 – C1-C6-ряда – бензофеноны и ксантоны;
соединения С6 – C2-C6-ряда – стилбены и антрахиноны;
соединения С6 – C3-C6-ряда – флавоноиды и изофлавоноиды;
соединения С6 – C3 – С3 – C 6 -ряда – лигнаны и неолигнаны;
полимерные фенольные соединения – лигнины, танины, танииды, меланины.
Индивидуальные флавоноиды могут быть названы по трем номенклатурам. Тривиальная номенклатура используются широко, в ней иногда указывают группу флавоноида или вид растения, послужившего источником для данного соединения. Например, названия, заканчивающейся на «-етин» обычно обозначают флавонолы (кверцетин), соединение трицин извлечен из растения, принадлежащего к роду Triticum. По рациональной номенклатуре в основе идет название структурного скелета, такие как флавон или халкон, например, 3,5,7,3’4′-пентагидроксифлавон. Наконец, флавоноидам может быть дано систематическое химическое название, например, 3,4-дигидро-2-фенил-2H-1-бензопиран для флавана, но этот метод является громоздким и используется редко [1].
Большинство флавоноидов – твердые кристаллические вещества, окрашенные в желтый цвет (флавоны, флавонолы, халконы, ауроны) или бесцветные (катехины, лейкоантоцианидины, флаваноны, изофлавоны). Наиболее яркие оттенки свойственны антоцианам, которые придают растительным тканям красную, синюю или фиолетовую окраску. Гликозилированные формы, как правило, хорошо растворимы в воде, нерастворимы или малорастворимы в органических растворителях (хлороформ, эфир и др.). Агликоны хорошо растворяются в низших спиртах (метиловом и этиловом), ацетоне, этилацетате и в растворах щелочей.
О-гликозиды при действии разбавленных минеральных кислот и ферментов более или менее легко гидролизуются до агликона и углеводного остатка. С-гликозиды с трудом расщепляются лишь при действии крепких кислот (концентрированная хлористоводородная или уксусная кислоты) или их смесей при длительном нагревании.
Легко окисляются в присутствии кислорода, под действием света и щелочей катехины и лейкоантоцианидины, превращаясь в окрашенные соединения – продукты конденсации, вплоть до высокомолекулярных полимерных форм. Остальные флавоноиды более устойчивы к окислению.
В зависимости от структуры фенольные соединения имеют разную биологическую активность и играют важную роль в растительном организме. В первую очередь это защитные вещества против различного рода патогенных микроорганизмов, а так же против насекомых и травоядных животных. Кроме того, они определяют свойства древесины и коры, обеспечивают различную окраску лепестков цветков и других частей растения. В растениях фенольные соединения играют важную роль в некоторых промежуточных этапах процесса дыхания. Установлено, что некоторые фенольные соединения играют важную роль в фотосинтезе в качестве кофакторов. Они используются растениями как энергический материал для разнообразных процессов жизнедеятельности, являются регуляторами роста, развития и репродукции, оказывая при этом как стимулирующее, так и ингибирующее воздействие.
Фенольные соединения имеют большое практическое значение для человека, обладая широким спектром фармакологического действия, при этом сила проявляемых ими эффектов зависит от многообразия их структур.
Токсичность фенольных соединений невелика и составляет следующий ряд, в котором первые два вещества практически безвредны: флавоноиды
Как осуществляется биосинтез фенольных соединений?
Хотя в обширную группу вторичных веществ фенольной природы входит более десяти классов различных по строению основного углеродного скелета природных соединений и каждый из этих классов объединяет сотни или даже тысячи (флавоноиды) индивидуальных соединений с существенными вариациями в природе прикрепленного к основному остову их молекулы набора заместителей (различия по числу и расположению в молекуле гидроксидных групп, остатков сахаров, органических кислот и других заместителей и т.п.), подавляющее большинство растительных фенольных соединений связано биогенетическим родством. Они составляют одно большое семейство веществ единого метаболического происхождения. Обусловлено это тем, что основной структурный элемент всех фенольных соединений – бензольное кольцо – образуется в растениях, как правило, по так называемому шикиматному пути. Синтезированный таким образом фрагмент ароматической структуры является той базовой единицей, из которой путем разных дополнительных превращений образуются почти все фенольные соединения растений. Лишь у ограниченного числа растительных фенолов ароматические кольца синтезируются по другому механизму – путем поликетидной конденсации ацетатных единиц.
Исходными компонентами в формировании ароматического ядра по шикиматному пути (схема 1) являются фосфоенолпируват (1), образующийся при гликолитическом распаде глюкозы, и эритрозо-4-фосфат (2) – промежуточный продукт окисления глюкозы по пентозофосфатному пути. При их конденсации образуется семиуглеродное соединение 7-фосфо-3-дезокси-D-арабиногептуло-зоновая кислота (3), которое затем подвергается циклизации, превращаясь в 3-дегидрохинную кислоту (4). На следующей стадии 3-дегидрохинная кислота теряет воду и превращается в 3-дегидрошикимовую кислоту (5) и далее под влиянием фермента оксидоредуктазы – в шикимовую кислоту (6), одно из важнейших промежуточных соединений пути, за что тот и получил свое название.
Шикимовая кислота по структуре близка ароматическим соединениям, однако ее шестичленное углеродное кольцо содержит только одну двойную связь. Дальнейшие преобразования этого кольца начинаются с фосфорилирования шикимовой кислоты по 3-му углеродному атому (7), а затем к фосфорилированной кислоте присоединяется молекула фосфоенолпирувата – получается 5-енолпирувилшикимат-3-фосфат (8). Последнее соединение претерпевает далее дефосфорилирование и дегидратацию, что приводит к образованию хоризмовой кислоты (9) – другого важного промежуточного соединения, которое в своем кольце имеет уже две двойные связи.
На этой стадии происходит разветвление шикиматного пути. По одному направлению из хоризмовой кислоты образуется L-триптофан (и далее индольные производные), по другому – L-фенилаланин и L-тирозин. Именно с последним ответвлением сопряжены дальнейшие превращения, которые в конечном счете приводят к образованию в растительных клетках фенольных соединений.
Начинается этот процесс с превращения хоризмовой кислоты превращается в префеновую кислоту (10). Последняя подвергается либо дегидратации, сопровождающейся декарбоксилированием, либо окислительному декарбоксилированию. В первом случае из префеновой кислоты образуется фенилпировиноградная (11), в другом – n-гидроксифенилпировиноградная кислота (13). Далее следует аминирование этих кетокислот с образованием соответственно L-фенилаланина (12) и L-тирозина (14).
Однако указанные трансформации могут совершиться и в другой последовательности. Аминирование может иметь место уже на стадии префеновой кислоты с преобразованием ее сначала в L-арогенную кислоту (15). Лишь затем молекула подвергается дегидратации с декарбоксилированием или окислительному декарбоксилированию, в результате которых образуются L-фенилаланин и L-тирозин.
Формированием этих двух ароматических аминокислот построение бензольного кольца завершается. Заканчивается и весь шикиматный путь, который как источник указанных аминокислот фактически представляет собой одну из составных частей первичного метаболизма клетки. Специфические вторичные превращения, ведущие к биосинтезу фенольных соединений, начинаются только после этой стадии метаболизма, причем они берут начало от одного-единственного продукта шикиматного пути – L-фенилаланина.
Первой, ключевой, реакцией на этом ответвлении вторичных превращений является реакция дезаминирования L-фенилаланина, катализируемая ферментом L-фенилаланин-аммиак-лиазой (схема 2).
В результате из L-фенилаланина (1) образуется транс-коричная кислота (2), которая на следующей стадии подвергается пара-гидроксилированию с образованием из нее n-гидроксикоричной (n-кумаровой) кислоты (3).
Пара-кумаровая кислота является первым и с биогенетической точки зрения простейшим фенольным соединением растений, которое служит родоначальником большинства других растительных фенолов. Она активизируется в КоА-лигазной реакции, а затем в виде активного КоА-эфира может вступать в реакции с различными другими метаболитами клетки или же подвергаться иным формам преобразований.
В результате таких превращений в растениях в виде уже конечных продуктов образуются представители разных классов полифенольных соединений. При окислительном укорачивании боковой цепи n-кумаровой кислоты образуются ацетофеноны, фенилуксусные, фенолкарбоновые кислоты. Восстановление ее боковой цепи вместе с последующей димеризацией или полимеризацией восстановленного продукта ведет к образованию лигнинов и полимерных фенолов типа лигнина. После введения дополнительной гидроксигруппы в орто-положении к боковой цепи происходит спонтанная циклизации последней с образованием кумаринов. Когда же n-кумаровая кислота подвергается этерификации или связывается с разными полимерными веществами клетки, то из нее образуются различные конъюгированные формы гидроксикоричных кислот и их производных.
Однако важнейшим ответвлением в комплексе возможных превращений n-кумаровой кислоты в фенольные соединения является путь, ведущий к образованию флавоноидов. На этом пути активированная n-кумаровая кислота последовательно вступает в реакцию с тремя молекулами активированной малоновой кислоты – малонил-КоА (схема 3).
В итоге к алифатической боковой цепочке этой кислоты по поликетидному типу конденсации углеродных единиц присоединяются три ацетатных фрагмента, из которых после внутримолекулярного замыкания (с участием фермента халконсинтазы) возникает второе бензольное кольцо 15-углеродного скелета флавоноидов. При этом сначала на основе такой структуры образуется халкон (1) – простейшая форма флавоноидов, у которой центральное гетероциклическое кольцо еще не замкнуто. Халкон же под влиянием соответствующей изомеразы обычно сразу превращается в свою изомерную форму – флаванон (2). Последний уже полностью обладает той типичной трехкольцевой структурой, которая характерна для большинства флавоноидов.
Так, существенной отличительной особенностью строения флавоноидов по сравнению со строением других полифенолов является двоякое биогенетическое происхождение двух бензольных колец их структуры. Одно из них синтезируется по шикиматному пути и является, таким образом, продуктом вторичных превращений аминокислоты L-фенилаланина. Другое же бензольное кольцо образуется по поликетидному механизму формирования углеродного скелета и получает свое начало от простейших продуктов обмена сахаридов.
Следует добавить, что образование структуры типа 5,7,4′-три-гидроксифлаванона или нарингенина является обязательной промежуточной стадией на пути биосинтеза всех флавоноидов. В дальнейшем могут происходить окислительные или восстановительные превращения, ведущие к изменению степени окисленности центрального гетероциклического кольца молекулы. В результате из нарингенина образуются все остальные классы флавоноидов: флавоны (3), флавонолы (4), антоцианидины (5), катехины – флаван-3-олы (6), флаван-3,4-диолы (7), изофлавоноиды и др.
Такие модификации идут по самостоятельным параллельным путям, причем их конечные продукты в виде представителей различных классов флавоноидов уже не подвергаются более поздним перестройкам основной структуры и взаимопревращениям. Теоретически помимо L-фенилаланина исходным предшественником синтеза полифенольных соединений по тому же пути может служить и другой конечный продукт шикииматного пути – ароматическая аминокислота L-тирозин. Однако активность соответствующего дезаминирующего фермента тирозин-аммиаклиазы чрезвычайно низка или вообще не обнаруживается в растениях, поэтому L-тирозин для биосинтеза полифенолов практического значения не имеет. Лишь у злаков он может играть некоторую дополнительную роль в качестве предшественника этих вторичных метаболитов. Отсюда следует, что подавляющее большинство всех фенолов растений фактически представляет собой большую семью родственных продуктов вторичного метаболизма L-фенилаланина, а пути их образования – общую систему параллельных ответвлений разных вторичных превращений этой ароматической аминокислоты.
В эту общую семью не входит только ограниченное число растительных фенолов. Так, в отдельных случаях n-гидроксибензойная и салициловая кислоты могут образовываться непосредственно из хоризмовой кислоты – одного из промежуточных продуктов шикиматного пути (см. схему 1). У некоторых растений (Rhus typhina, Camellia sinensis, Vaccinium vitis-idaea) прямой ароматизации, минуя стадию L-фенилаланина, может подвергаться и шикимовая кислота с образованием галловой кислоты. У этих растений, следовательно, и фенольная часть гидролизуемых дубильных веществ (которая построена из остатков галловой кислоты) может быть синтезирована непосредственно из шикимовой кислоты, а не из L-фенилаланина по стандартному пути биосинтеза фенольных соединений (схема 4).
Шикимовая кислота (1) почти всегда служит предшественником при биосинтезе производных нафтохинона. Вторым компонентом в этом биосинтезе является a-кетоглутаровая кислота (2), а важным промежуточным продуктом ее конденсации с шикимовой кислотой – о-сукцинилбензойная кислота (3). Далее следует циклизация с образованием уже типичных нафтохиноновых структур, где ароматическое кольцо построено на базе шикимовой кислоты, хиноидная же часть молекулы – из некарбоксильных С-атомов a-кетоглутаровой кислоты. Это нафтохинон-2-карбоновая кислота (4), нафтохинон (5).
У представителей семейства Rubiaceae сходным путем образуются и антрахиноновые производные. Дополнительное шестичленное углеродное кольцо их молекулы синтезируется путем конденсации нафтохинонового производного с диметилаллильной формой «активированного изопрена» – изопентенилдифосфата (ИПФФ). Продукт конденсации – диметилаллилнафтохинон (6), подвергаясь окислительной циклизации, превращается в антрахинон (7).
У других же высших растений антрахиноновые производные образуются из ацетатных-малонатных остатков по типу поликетидного синтеза. Антрахиноны являются, пожалуй, единственной группой растительных полифенолов, углеродный скелет которых целиком синтезируется по ацетатно-малонатному пути (схема 5).
В этом процессе в качестве молекулы-«затравки» участвует молекула ацетил-КоА (1), к которой последовательно присоединяются семь молекул малонил-КоА (2) с отщеплением от последних в ходе конденсации свободной карбоксильной группы и с образованием поликетидной цепи типа поликетокислоты (3). Эта кислота неустойчива и приобретает стабильную форму лишь после замыкания колец с образованием из нее промежуточного соединения – антрона (4 – кетоформа, 5 – енольная форма). Отличительной особенностью структуры антрона является наличие во 2-м положении его молекулы карбоксильной, а в 3-м – метильной групп. В ходе дальнейших реакций на пути биосинтеза антрахинонов и других антраценовых производных карбоксильная группа обычно отщепляется, а метильная либо сохраняется, либо окисляется в спиртовую или карбоксильную (б – эмодинантрон). Простейшим антрахиноновым производным является эмодин (7), который встречается почти во всех растениях, содержащих фенольные соединения типа антрахинонов.
Из других растительных фенолов по поликетидному механизму конденсации ацетатных единиц синтезируются лишь отдельные специфические соединения. К числу последних относятся, например, 6-метилсалициловая кислота и орселлиновая кислота, которые в основном встречаются в лишайниках.
Образовавшиеся фенолы всех основных классов и подклассов могут в дальнейшем подвергаться дополнительному окислению с увеличением числа фенольных ОН-групп в их молекуле. Через эти группы легко могут происходить реакции метилирования, гликозилирования и ацилирования, ведущие к включению разных заместителей в молекулу. Большинство фенолов встречается в растениях в форме водорастворимых гликозидов. Возможны и некоторые другие формы вторичной модификации основной структуры фенолов. В результате конечная структура индивидуальных соединений в пределах каждого класса фенолов может в широких пределах варьировать как по набору заместителей, так и по другим особенностям. Какими именно окажутся вторичные признаки строения у индивидуальных представителей полифенолов в каждом отдельном случае, определяет состав комплекса ферментов (метил-, гликозил- и ацилтрансфераз и др.) у конкретных видов растений.
Биосинтез фенольных соединений
БИОСИНТЕЗ ФЕНОЛЬНЫХ СОЕДИНЕНИЙ. К настоящее время выявлены 2 основных пути образования фенольных соединений: через шикимовую кислоту (шикиматный) и ацетатно-малонатный. Исходными соединениями биосинтез фенольных соединений по шикиматному пути служат фосфоенолпировиноградная кислота (фосфоенолпируват) и эритрозо-4-фосфат, образующиеся соответственно при гликолизе и в пентозо-фосфатном цикле (ПФЦ) при фотосинтезе. При их конденсации возникает семиуглеродное соединение — 2-кето-3-дезокси-7-фосфоарабогептоновая кислота Фермент синтетаза осуществляет циклизацию кислоты в 5-дигидро-хинную кислоту (I), которая способна затем превращаться в хинную кислоту либо — после дегидратации — в 5-дегидро-шикимовую кислоту (II). Последняя в присутствии фермента редуктазы (схема 1) восстанавливается в шикимовую кислоту (III).
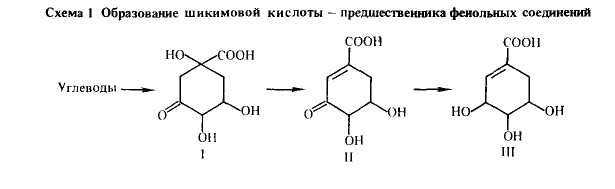
Шикимовая кислота имеет шестичленное кольцо, одну двойную связь, и ее довольно легко перевести в соединения ароматического ряда. Из нее возможно образование простых фенольных соединений С6-С1-ряда, например п-оксибензойной, протокатеховой и галловой кислот, а в дальнейшем и дубильных веществ гидролизуемой группы (схема 2).

Однако в растительной и микробной клетке превращение шикимовой кислоты в ароматические соединения идет значительно сложнее; процесс этот многоступенчатый и протекает с участием АТР с образованием 5-фосфо-шикимовой кислоты (IV), а затем через несколько стадий получается неустойчивое соединение — префеновая кислота (V). На стадии префеновой кислоты пути биосинтеза расходятся. По первому пути идет синтез фенилпировиноградной кислоты (VI), а по другому — п-оксипировиноградной кислоты (VII). При аминировании двух последних веществ образуются фенилаланин (VIII) и L-тирозин (IX). Данные аминокислоты могут участвовать в биосинтезе молекул белков и некоторых групп алкалоидов при дезаминировании аминокислот в присутствии ферментов — аммонийлиаз получаются транс-коричная и транс-гидроксикоричная кислоты (X и XI).
Из коричных кислот с помощью гидроксилирующих и метоксилирующих ферментов синтезируются соединения фенилпропанового ряда — оксикоричные кислоты (например, кофейная, феруловая, синаповая) и кумарины
Второй путь — ацетатно-малонатный — связан с промежуточным синтезом поликетометиленовых (по-ликетидных) предшественников Исходный продукт — ацетил-СоА — образуется в результате гликолиза Сахаров и содержит макроэргическую тиоэфирную связь. Ацетил-СоА при участии карбоксилазы и АТР в присутствии ионов Mn 2+ превращается в малонил-ацетил-СоА. Таким путем при постепенном наращивании углеродной цепи возникает поли-(3-кетометиленовая цепочка. Циклизация поликетидной цепи приводит к образованию различных фенольных соединений. Так, циклизация по С1 и С6-атомам приведет к синтезу производных флороглюцина, а циклизация по C2 — С7-атомам — к возникновению производных орселлиновой кислоты, которая является исходным продуктом в биогенезе лишайниковых кислот. Ацетатно-малонатный путь биосинтеза фенольных соединений широко распространен у грибов, лишайников и микроорганизмов. У высших растенийон обычно реализуется в сочетании с шикиматным путем в биосинтезе Флавоноидов и антрахинонов. Синтез флавоноидных соединений —
характерная особенность высших растений. Опыты с меченными по углероду С14 продуктами показали, что фенилпропановый скелет (кольцо B и трехуглеродный фрагмент) происходит от n-кумаровой кислоты, которая получается шикиматным путем. С другой стороны, кольцо А синтезируется по ацетатно-малонатному пути из трех молекул малонил-СоА
В результате взаимодействия п-кумароил-СоА с 3 молекулами малонил-СоА образуется халкон или флаванон (схема 3).
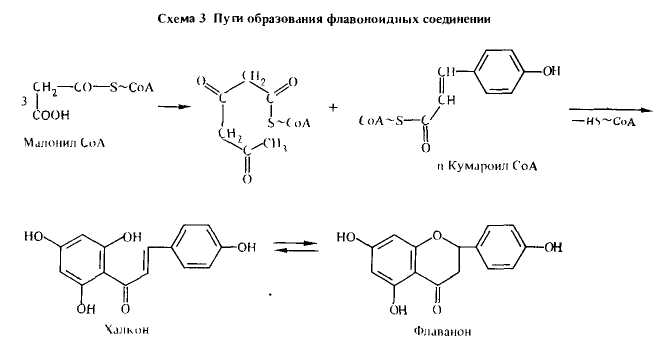
Природа первичного продукта циклизации пока не уточнена, что объясняется легкостью взаимопревращений халконов и флаванонов. Из последних образуются все остальные группы флавоноидов.
О биосинтезе лигнанов известно немного. Возможный путь их биосинтеза — восстановительная конденсация двух С6-С3-единиц с ненасыщенными боковыми цепями.
Биосинтез некоторых фенолов помимо двух основных путей осуществляется иначе. Некоторые ароматические соединения изопреноидной структуры, являющиеся компонентами эфирномасличных растений (тимол, карвакрол, эвгенол и др.), образуются из мевалоновой кислоты через диметилаллилпирофосфат с последующей стадией ароматизации шестичленных колец.
Источники:
http://studwood.ru/1643588/matematika_himiya_fizika/harakteristika_fenolnyh_soedineniy
http://www.pharmspravka.ru/farmatsevticheskie-vorosyi-i-otvetyi/kak/kak-osuschestvlyaetsya-biosintez-fenolnyih-soedin.html
http://www.9lc.com/biosintez-fenolnih-soedineniy.html





