
Секвенирование генома винограда
Секвенирование генома винограда
РФФИ поддержал научное исследование аспиранта ВИР по секвенированию генома дикорастущего вида винограда, иммунного к грибным заболеваниям
 Аспирант ВИР Магамедгусейн Агаханов (научный руководитель – доктор биологических наук Е. К. Потокина) получил поддержку Российского фонда фундаментальных исследований (РФФИ) на осуществление секвенирования генома дикорастущего вида винограда, устойчивого к грибным патогенам.
Аспирант ВИР Магамедгусейн Агаханов (научный руководитель – доктор биологических наук Е. К. Потокина) получил поддержку Российского фонда фундаментальных исследований (РФФИ) на осуществление секвенирования генома дикорастущего вида винограда, устойчивого к грибным патогенам.
По условиям гранта РФФИ, исследование ляжет в основу диссертации аспиранта на соискание ученой степени кандидата наук. Проект должен быть реализован за два года, его выполнение начнется 1 октября 2019 г.
Полное название проекта, выигравшего конкурс РФФИ, – “Полногеномное секвенирование иммунного к грибным заболеваниям вида Vitis rotundifolia Michx. для выявления интрогрессий в геномах отдаленных гибридов, полученных с его участием”. Как пишет Магамедгусейн в аннотации, создание новых сортов культурного винограда с групповой устойчивостью к грибным болезням и филлоксере на основе отдаленной гибридизации с участием иммунного вида V. rotundifolia Мichx. вызывает большой интерес. Однако получить гибриды между видом V. vinifera (подрод Euvitis, 38 хромосом) и видом V. rotundifolia Michaux (подрод Muscadinia, 40 хромосом) ранее удалось с большим трудом, при этом гибридные сеянцы становились фертильными только после их полиплоидизации (аллотетраплоидии). На сегодняшний день в НИИ виноградарства и вина “Магарач” в результате сотрудничества с селекционерами разных стран и собственных экспериментов по отдаленной гибридизации создан новый уникальный генофонд отдаленных гибридов, несущих в своем геноме гены Vitis rotundifolia Michx., что позволяет изучать механизмы устойчивости к грибным болезням. Так как на сегодняшний день информация о геноме Vitis rotundifolia отсутствует, то достаточно трудно выявить интрогрессии его генома у полученных отдаленных гибридов.
В коллекции ВИР сохраняются аутентичные образцы Vitis rotundifolia, и цель проекта заключается в проведении исследований, направленных на полногеномное секвенирование этих образцов Vitis rotundifolia Michx. с использованием нанопор MinION (Oxford Technologies). В результате появится возможность выявлять в геномах отдаленных гибридов винограда фрагменты хромосом, интрогрессированных от это иммунного к грибным заболеваниям вида. В задачи проекта входит также получение родительских форм V. vinifera и V.rotundifolia с признаками полиплоидов путем обработки колхицином меристемных тканей почек для последующего проведения экспериментов по отдаленной гибридизации.
Предполагается, что по результатам первого этапа реализации гранта РФФИ Магамедгусейн Агаханов подготовит не менее одной статьи для публикации в журнале, индексируемом в международных базах данных, и выступит на научном мероприятии с докладом о своем исследовании. А по окончании второго этапа будет опубликовано не менее двух статей в журналах, индексируемых в международных базах данных. В течение года после окончания второго этапа диссертация Магамедгусейн Агаханова должна быть принята к защите диссертационным советом.
Научный руководитель проекта – доктор биологических наук Елена Потокина отметила, что поддержка РФФИ подтвердила актуальность исследования аспиранта и новизну ожидаемых результатов.
Магамедгусейн Агаханов стал одним из 1,5 тысяч молодых ученых России, выигравших конкурс на поддержку лучших проектов фундаментальных научных исследований, проводившийся РФФИ в рамках проекта “Наука”.
Напомним, что в июле 2019г. научные сотрудники ВИР привлекли внимание школьников к этой теме – расшифровка генома дикорастущего предка культурного винограда стала одной из двух тем, по которой работали старшеклассники в образовательном центре “Сириус”.
Секвенирование генома винограда

Предпосылками для формирования генетики как самостоятельной научной области послужило открытие законов Менделя. В дальнейшем в XX веке было сделано четыре открытия, положивших начало развитию генетики [1]:
• установлены клеточные основы наследственности — хромосомы;
• определена молекулярная основа наследственности — двойная спираль ДНК;
• открыта информационная основа наследственности, а также биологический механизм, с помощью которого клетки считывают информацию, содержащуюся в генах;
• изобретены технологии клонирования и секвенирования рекомбинантных ДНК.
Последняя четверть прошлого века была отмечена неустанным стремлением расшифровать сначала гены, а затем и целые геномы [2].
Первая рабочая концепция секвенирования — метод Сэнгера, также известный как метод обрыва цепи, — была предложена в 1977 году. За это открытие Фредерик Сэнгер был удостоен Нобелевской премии по химии в 1980 году. Этот метод секвенирования применялся в течение 40 лет, а его усовершенствование и коммерциализация привели к широкому распространению секвенирования [2].
Описание метода Сэнгера
Секвенирование Сэнгера — метод, при котором используются олигонуклеотидные праймеры для поиска определенных областей ДНК. Этот процесс начинается с деспирализации двухцепочечной ДНК [5]. Одна цепочка ДНК является матрицей для синтеза комплементарной цепочки при помощи фермента ДНК-полимеразы. Реакцию с одной и той же цепочкой проводят в четырех разных пробирках, каждая из которых содержит [3]:
— праймер;
— четыре дезоксинуклеотида (дезоксиаденозинтрифосфат, дезоксигуанозинтрифосфат, дезоксицитидинтрифосфат и тимидинтрифосфат);
— небольшое количество (1 к 100) одного из радиоактивно меченных дезоксинуклеотидов (для визуализации продуктов реакции).
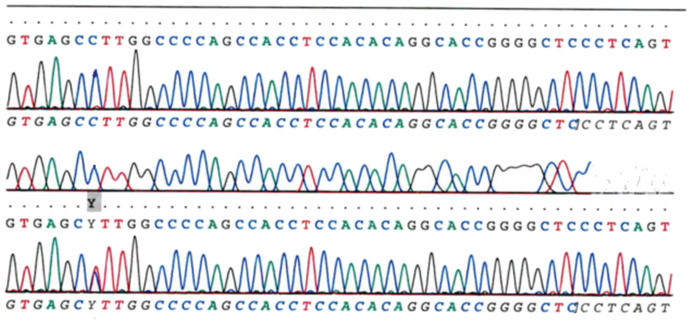
В каждой пробирке образуется набор фрагментов ДНК разной длины, заканчивающихся одним и тем же нуклеотидом. После завершения реакции содержимое пробирок разделяют электрофорезом в полиакриламидном геле в денатурирующих условиях и затем проводят авторадиографию гелей. Каждый дезоксинуклеотид отмечен флуоресцентным маркером: A — зеленый цвет, T — красный, G — черный и C — синий. Лазер в автомате, используемый для считывания последовательности, фиксирует интенсивность флуоресценции. Когда в последовательности встречается гетерозиготный вариант, локусы захватываются двумя флуоресцентными красителями одинаковой интенсивности. Если присутствует гомозиготный вариант, ожидаемый флуоресцентный цвет заменяется цветом комплементарного основания [5].
Продукты четырех реакций формируют «секвенирующую лестницу», которая позволяет «прочитать» нуклеотидную последовательность фрагмента ДНК. Метод Сэнгера позволяет также определять нуклеотидную последовательность РНК, но она предварительно должна быть «переписана» в формат ДНК с помощью обратной транскрипции [3].
Секвенирование Сэнгера — это надежный метод для определения генных мутаций, который широко использовался в течение нескольких десятилетий. Метод Сэнгера является геноспецифичным, и с его помощью анализируют небольшое подмножество генов. Секвенирование Сэнгера позволяет идентифицировать мозаичные мутации. Но метод секвенирования Сэнгера не позволяет проводить точную количественную оценку, то есть нельзя сделать вывод о том, в каком количестве клеток есть мутация [5].
Метод дробовика
Метод дробовика используется для секвенирования длинных участков ДНК. Суть метода состоит в получении случайной массированной выборки клонированных фрагментов ДНК данного организма, на основе которых восстанавливается исходная последовательность ДНК [6].
Первые методы секвенирования способны восстанавливать небольшие последовательности ДНК (порядка 1000 нуклеотидов), следовательно, для секвенирования более длинных последовательностей требовалось разработать новый подход. При секвенировании методом дробовика ДНК случайным образом фрагментируется на мелкие участки с помощью сайт-неспецифичных нуклеаз. Затем фрагменты секвенируют любым доступным методом, например, методом секвенирования по Сэнгеру. Полученные перекрывающиеся случайные фрагменты ДНК собирают с помощью специального программного обеспечения в одну целую последовательность. Данный метод оставался фундаментальным методом секвенирования генома в течение 20 лет [2]. В 1981 году метод применен на практике — полное секвенирование генома вируса мозаики цветной капусты [7].
На практике трудности возникают из-за повторяющихся последовательностей. Например, можно легко секвенировать типичные бактериальные геномы (около 1,5 % повторов) или эухроматическую часть генома мухи (около 3 % повторов). Человеческий геном содержит более чем 50 % повторяющихся последовательностей. Такие особенности усложняют сборку правильной и законченной последовательности генома [2].
В дальнейшем этот подход совершенствовался: были улучшены механизмы фрагментации и клонирования ДНК. В 1990 году был предложен метод секвенирования парных прочтений. Результаты первого применения метода секвенирования парных концов на практике были опубликованы в 1990 году в работе, посвященной секвенированию человеческого гена гипоксантин-гуанинфосфорибозилтрансферазы [4].
При секвенировании парных прочтений ДНК разрезается на случайные фрагменты, которые затем группируются по весу и клонируются в векторах. Клоны секвенируют с обоих концов с использованием метода обрыва цепи, в результате которого образуются две коротких последовательности [4].
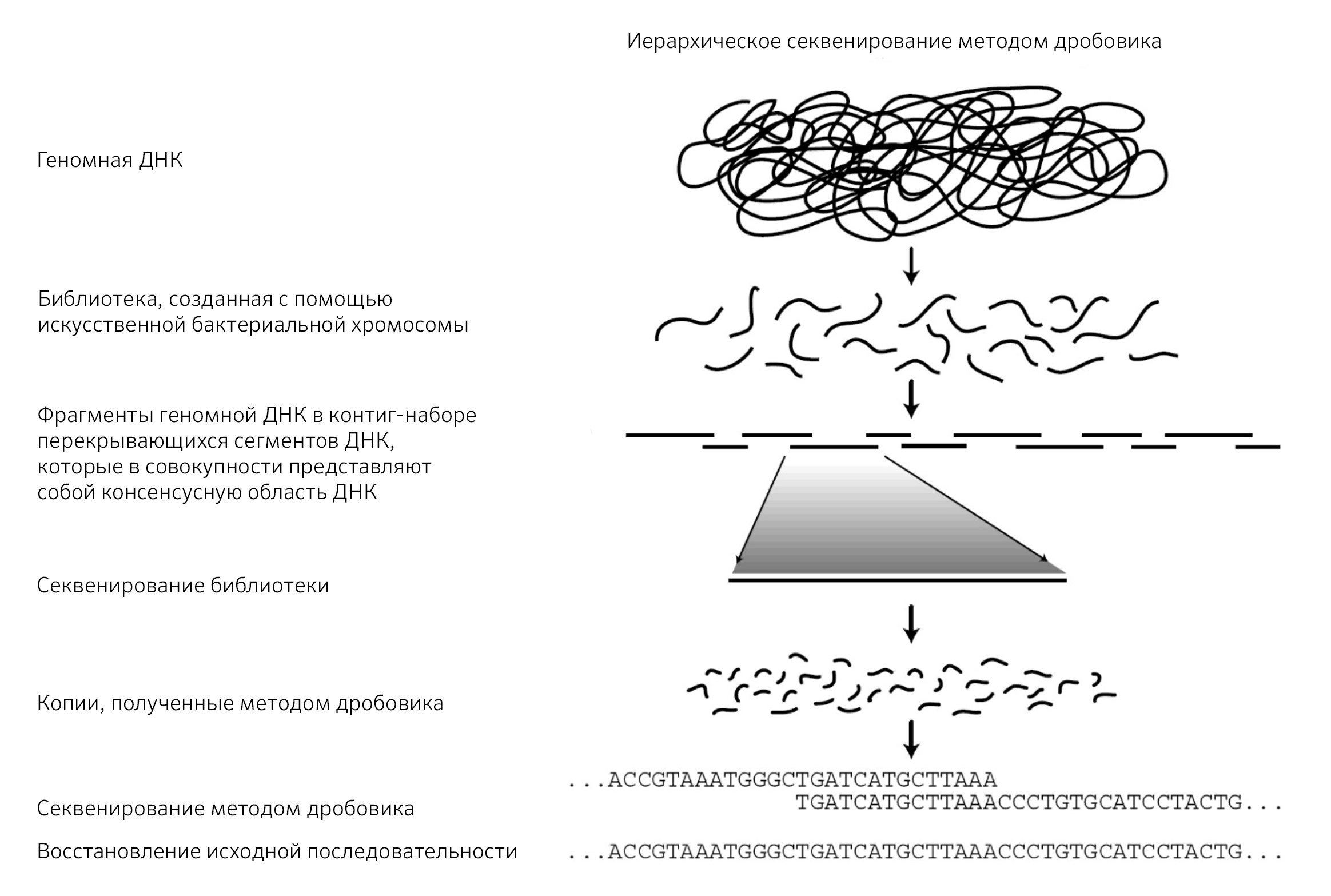
Иерархическое секвенирование методом дробовика
Для секвенирования больших геномов, содержащих повторяющиеся последовательности, используется подход «иерархического секвенирования методом дробовика» [2].
Данный метод — технически более сложный из-за необходимости обработки больших объемов данных. Это служит причиной тому, что метод иерархического секвенирования имеет более высокую стоимость [2].
Переломной точкой развития методов секвенирования стало появление и широкое распространение технологий ПЦР, автоматизация этапов «чтения» ДНК, совершенствование программного обеспечения. Все это дало начало созданию методов секвенирования следующего поколения. Секвенаторы нового поколения значительно дешевле и гораздо эффективнее своих предшественников. На сегодняшний день производительность некоторых секвенаторов измеряется уже сотнями миллиардов пар оснований, что, например, позволяет подобным приборам сканировать индивидуальный геном человека всего за несколько дней.
Источники:
- International Human Genome Sequencing Consortium et al. Initial sequencing and analysis of the human genome //nature. – 2001. — Т. 409. – №. 6822. – С. 860.
- Pareek C. S., Smoczynski R., Tretyn A. Sequencing technologies and genome sequencing //Journal of applied genetics. – 2011. – Т. 52. – №. 4. – С. 413-435.
- Egli M. et al. Nucleic acids in chemistry and biology. – Royal Society of Chemistry, 2006.
- Edwards A. et al. Automated DNA sequencing of the human HPRT locus //Genomics. – 1990. – Т. 6. – №. 4. – С. 593-608.
- Solomon D. A. Integrating Molecular Diagnostics With Surgical Neuropathology //Practical Surgical Neuropathology: A Diagnostic Approach. – Elsevier, 2018. – С. 71-89.
- Staden R. A strategy of DNA sequencing employing computer programs //Nucleic acids research. – 1979. – Т. 6. – №. 7. – С. 2601-2610.
- Gardner R. C. et al. The complete nucleotide sequence of an infectious clone of cauliflower mosaic virus by M13mp7 shotgun sequencing //Nucleic acids research. – 1981. – Т. 9. – №. 12. – С. 2871-2888.
Полное секвенирование генома позволит лечить рак индивидуально

Полное секвенирование генома злокачественного новообразования поможет разработать индивидуальный подход к лечению каждого пациента.
Человеческий геном “прописан” в молекуле ДНК в виде “букв” – блоков, которые учёные называют нуклеотидами. Каждый такой “кирпичик” имеет своё международное название, которое для удобства генетики сокращают до первой буквы.
Так, эти органические соединения обозначаются буквами A (аденин), C (цитозин), G (гуанин) и T (тимин). Обычно нуклеотиды группируются в пары строго определённым образом. Однако иногда “правописание” ДНК допускает ошибки и, например, на место A становится G.
Эти изменения или мутации могут быть спонтанными или возникать под влиянием неблагоприятных факторов. Так, виновником “поломки” может стать и табачный дым, и ультрафиолетовое излучение, и различные химические вещества. Каждая мутация оставляет определённый отпечаток в ДНК.
Клетки, делясь и размножаясь, воспроизводят точные копии своей ДНК, поэтому любые “орфографические ошибки” в ней будут в точности повторены и переданы дальше. Со временем количество таких неполадок накапливается. Это может приводить в том числе к неконтролируемому росту клеток, другими словами, развитию опухоли.
Группа учёных из Кембриджского университета, работающая под руководством Серены Ник-Зайнал (Serena Nik-Zainal), предложила использовать для борьбы с такими заболеваниями секвенирование генома раковой опухоли.
Этот метод позволяет “прочесть” всю генетическую схему раковой клетки. Если сравнить полученные данные с геномом здоровых клеток заболевшего человека, можно увидеть, как именно мутировала ДНК и, исходя из этого, разработать оптимальную стратегию лечения.
Чтобы выяснить, насколько применим и полезен такой метод в повседневной клинической практике, исследователи из Кембриджа объединились с коллегами из Швеции.
Дело в том, что шведские онкологи проводят общенациональный проект под названием SCAN-B, в котором с 2010 года участвуют все женщины, заболевшие раком молочной железы. Соответственно, медики собрали большую базу данных результатов обследований и лечения своих пациенток.
Воспользовавшись этими обширными данными, исследователи выполнили полное секвенирование генома опухолевых клеток 254 женщин, у которых был обнаружен так называемый тройной негативный рак молочной железы.
Этот вариант болезни учёные выбрали неспроста. Тройной негативный рак – это одна из наиболее агрессивных опухолей с самым плохим прогнозом выживаемости.
Своё название недуг получил потому, что у таких образований отсутствуют три основные молекулы-мишени (рецепторы), на которые обычно воздействуют лекарства.
Закончив секвенирование, исследователи применили алгоритм машинного обучения под названием HRDetect, который генетики разработали ранее для идентификации опухолей с характерными “генетическими подписями”, вызванными мутациями в генах BRCA1 или BRCA2.
Стоит пояснить, что алгоритм был разработан именно для этих двух генов потому, что присутствие любого из них увеличивает риск развития рака молочной железы.
Таким образом, HRDetect может предсказать вероятность именно BRCA1/BRCA2 типа рака, для которого лучшим способом лечения на сегодня считается сравнительно новый класс препаратов – ингибиторы PARP.
Обработав данные секвенирования геномов опухолей пациенток, алгоритм разделил всех женщин на три группы, присвоив высокий, средний или низкий рейтинг.
Высокий предполагал, что женщины, вероятнее всего, имеют BRCA1/BRCA2-ассоциированную опухоль. Их отклик на приём существующих препаратов и прогнозы на выздоровление были самыми лучшими.
Удивительно, но группа, которой было присвоено промежуточное количество баллов и которая должна была бы сравнительно неплохо реагировать на современные лекарства, показала наихудший отклик на имеющиеся в арсенале онкологов средства.
Это означает, что опухоли у этих пациенток были, вероятно, спровоцированы отличными от BRCA1/BRCA2 мутациями и требовали других лечебных подходов.
И это весьма ценная информация. “Прочтение” генома опухолей пациенток этой категории дало специалистами подсказку, какие механизмы приводят к развитию их разновидности рака. А значит, экспертам будет проще разрабатывать новые эффективные лекарства и схемы лечения.
Последняя группа, которой искусственный интеллект по результатам секвенирования присвоил низкий рейтинг, тоже откликалась на лечение далеко не лучшим образом, то есть их рак также был вызван иными повреждениями ДНК.
Однако геном новообразований этих участниц указывал на биологические изменения, которые потенциально могут быть “атакованы” как уже существующими лекарствами, так и препаратами, проходящими клинические испытания. К ним относятся, например, ингибиторы иммунных контрольных точек (“Вести.Наука” подробно рассказывали об этом классе препаратов).
По словам исследователей, секвенирование генома опухолей помогает точно определить степень “откликаемости” рака на планируемое лечение. Особенно важно иметь такую информацию, приступая к лечению женщин с тройным негативным раком, который всё ещё считается трудноизлечимым заболеванием.
Но самым значимым, по мнению учёных, является тот факт, что секвенирование генома раковых клеток помогает выяснить, что же происходит в опухолях, плохо поддающихся существующей противораковой терапии. Понимание этого процесса поможет разработать новые эффективные способы побороть опасную болезнь.
Кстати, прогресс в технологиях секвенирования весьма впечатляет. Полный анализ генома опухоли сегодня можно провести всего за 24 часа. Ещё 24-48 часов онкологам необходимо для анализа полученных данных.
Поэтому уже сегодня вполне реально сделать секвенирование рутинным методом исследования, чтобы оценить течение болезни каждого пациента и разработать индивидуальный наиболее эффективный план лечения.
Предложенный метод, считают исследователи, открывает путь к избавлению даже от самых сложных и устойчивых к терапии злокачественных образований.
“Секвенирование даёт нам полное представление о геноме рака. Оно раскрывает многие вещи, которые мы не могли увидеть ранее, потому что мы их даже не искали. Полная “карта” ракового генома каждого пациента помогает понять, что вызвало опухоль в каждом конкретном случае и эффективнее лечить каждого конкретного человека”, – поясняет Серена Ник-Зайнал в пресс-релизе университета.
Результаты исследования международной команды учёных опубликованы в престижном научном издании Nature Medicine.
Напомним, что авторы “Вести.Наука” (nauka.vesti.ru) рассказывали и об эпигенетических факторах риска развития рака (к счастью, обратимых), о мутациях ДНК, “выдающих” рак на ранней стадии и о пользе определённых физических нагрузок при раке груди.
Чтобы всегда оставаться в курсе самых интересных научных новостей, подписывайтесь на наши группы в социальных сетях: ВК, Facebook, Twitter, “Одноклассники”. Есть мы и в Яндекс.Дзене.
Источники:
http://www.vir.nw.ru/blog/2019/08/26/rffi-podderzhal-nauchnoe-issledovanie-aspiranta-vir-po-sekvenirovaniyu-genoma-dikorastushhego-vida-vinograda-immunnogo-k-gribnym-zabolevaniyam/
http://medach.pro/post/2157
http://nauka.vesti.ru/article/1233018




